Basic UDI-DI (bUDI) - what is it, where to obtain it, and what to do with it
With the introduction of European regulations MDR and IVDR, governing the circulation of medical devices in the European Union, a set of requirements emerged, compliance with which is necessary for manufacturers wishing to distribute their medical products in Europe. Basic UDI-DI (bUDI) is one such requirement. In this article, we will explore what the basic UDI-DI entails, how it can be obtained, and how it should be utilised.
What is Basic UDI-DI (bUDI-DI)
Basic UDI-DI is the identifier of a medical device within the territory of the European Union. Its presence is one of the mandatory requirements of European legislation in the field of medical devices. It emerged after the implementation of European regulations MDR 2017/745 and IVDR 2017/746. The necessity to obtain and use the basic UDI-DI identifier is regulated by Article 29 of the MDR and Article 26 of the IVDR. According to these articles, every manufacturer of medical devices or in vitro diagnostic devices is obliged to obtain a basic UDI-DI in order to legally market their products in the EU.
More detailed requirements for the basic UDI-DI can be found in Sections C of Annexes VI to the MDR and IVDR. Additional information is available in the MDCG guidance 2018-1.
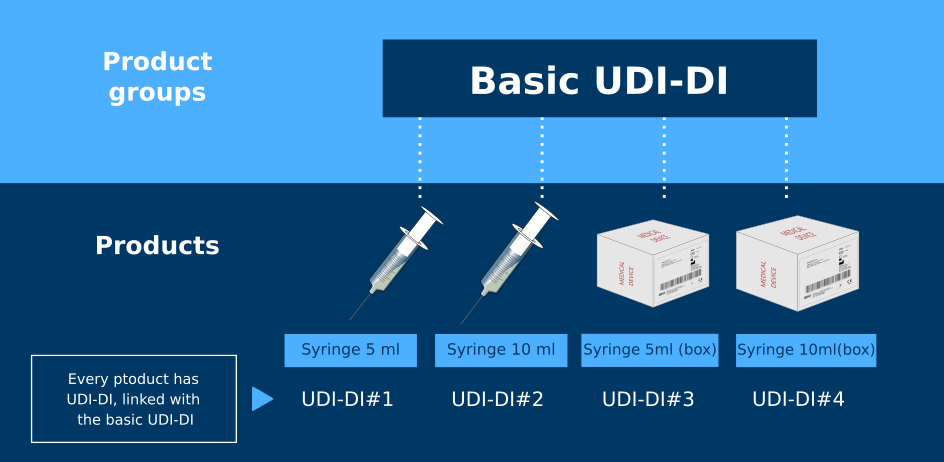
The basic UDI-DI is a unique identifier linked to a specific group of products from a single manufacturer. It's important not to confuse the basic UDI-DI with the regular UDI-DI (which does not contain the word "basic" at the beginning).
UDI-DI stands for Unique Device Identifier - Device Identifier (there is also PI - Production Identifier). It is a code used to identify individual medical devices or their packaging, accompanied by a product barcode. The UDI-DI system has been in use in the United States for a long time, and with the introduction of the MDR and IVDR, it has also come into effect in the European Union. It is important to understand that the Basic UDI-DI and the Common UDI-DI serve different purposes. The Basic UDI-DI is unique to Europe.
UDI and basic UDI
How to obtain a basic UDI-DI
At the core of the basic UDI is a company code (prefix) issued to the manufacturer by specialised organisations, which is used for encoding product barcodes.
There are four specialised organisations accredited by the European Union to issue such prefixes:
GS1
HIBCC (Health Industry Business Communications Council)
ICCBBA
IFA GmbH (Informationsstelle für Arzneispezialitäten)
To obtain a basic UDI-DI, it is necessary to use encoding standards developed by one of the specified organisations. To carry out this procedure, you should contact one of them.
What constitutes a basic UDI-DI
The basic UDI-DI is a alphanumeric code composed of several components. Let's break down the structure of a basic UDI-DI using an identifier formed in accordance with GS1 standards as an example.
Basic UDI (bUDI)
The first part of the basic UDI-DI, generated in accordance with GS1 standards, is the company prefix assigned by GS1 upon the company's registration in the GS1 electronic system. Next comes the code that the manufacturer assigns to a group of products using their own standards. The last two digits of the basic UDI-DI are generated using a special calculator designed for the generation and validation of basic UDI-DIs.
How to group products for assigning a basic UDI
A Basic UDI-DI can be assigned not to individual products, but to groups of products that share specific characteristics. Therefore, to assign a Basic UDI-DI, it is necessary first and foremost to group the products - each group will receive its own identifier. Product grouping is the responsibility of the manufacturer.
In order for products to be grouped together, they must meet the following criteria:
Intended purpose. Products in the same group must have the same or similar intended purpose, meaning they are designed for analogous medical aims or procedures.
Risk class. Products in the group must belong to the same risk class as determined by medical regulators.
Device type. Products must have a similar device type, indicating that their design and functionality are alike or share common features.
Manufacturing specifications. Products in the group must possess similar manufacturing characteristics, such as materials, production methods, or assembly processes.
Assigning basic UDI-DIs and their correct grouping is crucial for ensuring the identification and tracking of medical devices in the medical industry, as well as for compliance with regulatory authorities' requirements.
How the basic UDI is applied
The Basic UDI-DI is an important part of product documentation. It is indicated in:
Basic UDI-DI (bUDI) - what is it, where to obtain it, and what to do with it
With the introduction of European regulations MDR and IVDR, governing the circulation of medical devices in the European Union, a set of requirements emerged, compliance with which is necessary for manufacturers wishing to distribute their medical products in Europe. Basic UDI-DI (bUDI) is one such requirement. In this article, we will explore what the basic UDI-DI entails, how it can be obtained, and how it should be utilised.
What is Basic UDI-DI (bUDI-DI)
Basic UDI-DI is the identifier of a medical device within the territory of the European Union. Its presence is one of the mandatory requirements of European legislation in the field of medical devices. It emerged after the implementation of European regulations MDR 2017/745 and IVDR 2017/746. The necessity to obtain and use the basic UDI-DI identifier is regulated by Article 29 of the MDR and Article 26 of the IVDR. According to these articles, every manufacturer of medical devices or in vitro diagnostic devices is obliged to obtain a basic UDI-DI in order to legally market their products in the EU.
More detailed requirements for the basic UDI-DI can be found in Sections C of Annexes VI to the MDR and IVDR. Additional information is available in the MDCG guidance 2018-1.
The basic UDI-DI is a unique identifier linked to a specific group of products from a single manufacturer. It's important not to confuse the basic UDI-DI with the regular UDI-DI (which does not contain the word "basic" at the beginning).
UDI-DI stands for Unique Device Identifier - Device Identifier (there is also PI - Production Identifier). It is a code used to identify individual medical devices or their packaging, accompanied by a product barcode. The UDI-DI system has been in use in the United States for a long time, and with the introduction of the MDR and IVDR, it has also come into effect in the European Union. It is important to understand that the Basic UDI-DI and the Common UDI-DI serve different purposes. The Basic UDI-DI is unique to Europe.
UDI and basic UDI
How to obtain a basic UDI-DI
At the core of the basic UDI is a company code (prefix) issued to the manufacturer by specialised organisations, which is used for encoding product barcodes.
There are four specialised organisations accredited by the European Union to issue such prefixes:
GS1
HIBCC (Health Industry Business Communications Council)
ICCBBA
IFA GmbH (Informationsstelle für Arzneispezialitäten)
To obtain a basic UDI-DI, it is necessary to use encoding standards developed by one of the specified organisations. To carry out this procedure, you should contact one of them.
What constitutes a basic UDI-DI
The basic UDI-DI is a alphanumeric code composed of several components. Let's break down the structure of a basic UDI-DI using an identifier formed in accordance with GS1 standards as an example.
Basic UDI (bUDI)
The first part of the basic UDI-DI, generated in accordance with GS1 standards, is the company prefix assigned by GS1 upon the company's registration in the GS1 electronic system. Next comes the code that the manufacturer assigns to a group of products using their own standards. The last two digits of the basic UDI-DI are generated using a special calculator designed for the generation and validation of basic UDI-DIs.
How to group products for assigning a basic UDI
A Basic UDI-DI can be assigned not to individual products, but to groups of products that share specific characteristics. Therefore, to assign a Basic UDI-DI, it is necessary first and foremost to group the products - each group will receive its own identifier. Product grouping is the responsibility of the manufacturer.
In order for products to be grouped together, they must meet the following criteria:
Intended purpose. Products in the same group must have the same or similar intended purpose, meaning they are designed for analogous medical aims or procedures.
Risk class. Products in the group must belong to the same risk class as determined by medical regulators.
Device type. Products must have a similar device type, indicating that their design and functionality are alike or share common features.
Manufacturing specifications. Products in the group must possess similar manufacturing characteristics, such as materials, production methods, or assembly processes.
Assigning basic UDI-DIs and their correct grouping is crucial for ensuring the identification and tracking of medical devices in the medical industry, as well as for compliance with regulatory authorities' requirements.
How the basic UDI is applied
The Basic UDI-DI is an important part of product documentation. It is indicated in:
Certificates issued by notified bodies.
EU Declaration of Conformity.
Technical documentation.
Certificates of Free Sale.
Furthermore, the Basic UDI-DI is used when registering products in the EUDAMED system and when interacting with notified bodies.
We only use essential cookies that enable core functionality and proper operation of the website. These cookies do not store any personally identifiable data. By continuing to use this website, you consent to the use of the essential cookies. You may disable these cookies by changing your browser settings, but this may affect how the website functions. We do not use our own or third-party analytical, preferences, statistics, marketing, functional, advertisement, performance or any other non-essential cookies.